For this interview, I reached out to Janus Juul Eriksen, a Ph.D. fellow at Aarhus University in Denmark. Janus is a chemist by trade without any formal education in computer science; but he is getting up to 12x speed-up compared to his CPU-only code after modifying less than 100 lines of code with one week of programming effort.
For this interview, I reached out to Janus Juul Eriksen, a Ph.D. fellow at Aarhus University in Denmark. Janus is a chemist by trade without any formal education in computer science; but he is getting up to 12x speed-up compared to his CPU-only code after modifying less than 100 lines of code with one week of programming effort.
How did he do this? He used OpenACC.
OpenACC is a simple, powerful and portable approach for researchers and scientists who need to rapidly boost application performance for faster science while minimizing programming. With OpenACC, the original source code is kept intact, making the implementation intuitively transparent and leaving most of the hard work to the compiler.
NVIDIA recently announced the new OpenACC Toolkit, an all-in-one suite of parallel programming tools, that helps researchers and scientists quickly accelerate applications.
“OpenACC is much easier to learn than OpenMP or MPI. It makes GPU computing approachable for domain scientists,” says Janus. “Our initial OpenACC implementation required only minor efforts, and more importantly, no modifications of our existing CPU implementation.”
Janus is part of the research team developing the quantum chemistry code LSDalton, a massively parallel and linear-scaling program for the accurate determination of energies and other molecular properties for large molecular systems.
In need of speed, the LSDalton team was awarded an INCITE allocation which gave them access to Oak Ridge National Laboratory’s Titan supercomputer. With this, they needed to find a way to use the power of the supercomputer: enter OpenACC. Demonstrating success on Titan with their GPU-accelerated code, they were recently one of 13 application code projects selected to join the Center for Accelerated Application Readiness (CAAR) program. This means they will be among the first applications to run on Summit, the new supercomputer debuting in 2018 which will deliver more than five times the computational performance of Titan’s 18,688 nodes.
This access will enable the research team to simulate larger molecular structures at higher accuracy, ultimately accelerating discoveries in materials and quantum chemistry.
B: In layman’s terms, what is your team up to?
J: For the last three decades, it has become increasingly more common to interpret various macroscopic chemical phenomena and reaction mechanisms in terms of specific inter- and intramolecular interactions, (on a more microscopic scale). Nowadays, this is not only the case within the classical fields of physics and chemistry but also within more modern areas of natural sciences such as molecular biology and nanotechnology. Thus, quantum chemistry, that is, the application of quantum mechanics to molecular systems and phenomena, has become an integral tool to all of chemical, biological, and general material sciences.
Besides contributing qualitative information on molecules and their different interactions, modern quantum chemistry may also provide a deeper understanding of molecular processes, which cannot be derived from experimental work alone. In fact, the majority of experimental results are nowadays supported by modern computational work, and theory is more than ever used as an apparatus to lead and guide future experimental work within the medicinal industry and material sciences. As a result, accurate computer simulations on increasingly larger molecular systems are desired, not only by academia but also by various industrial research labs.
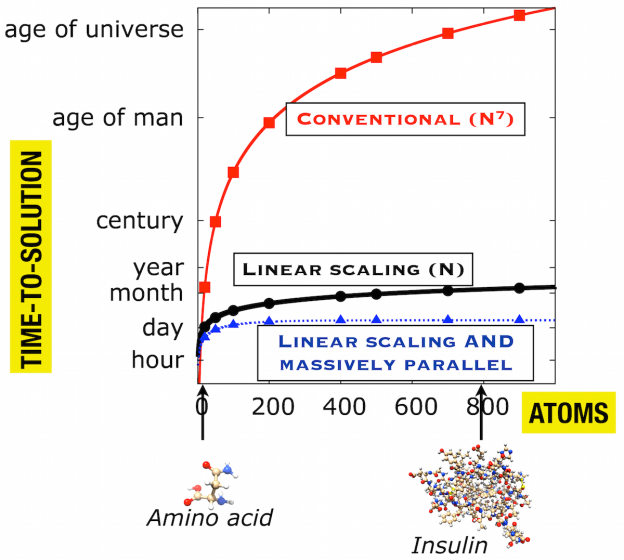
The hiccup, however, is the fact that the computational scaling on the size of the molecular system grows dramatically whenever an increasingly higher accuracy is requested. So, in order to circumvent this (unphysical) computational problem, so-called local correlation methods have been devised, which describe the fundamental repulsive interactions between individual electrons in a spatially local manner instead of the typical delocalized, canonical manner (See Figure 2).
B: What made you decide to develop on OpenACC?
J: One of the main requirements of the LS-Dalton code as a whole is it should be portable. The code itself has taken a radical turn towards HPC in recent years, through the development of massively parallel implementations of numerous quantum chemical methods. Portability remains of key importance, and the code is bound to install and run as easily on a regular workstation or a modest Beowulf cluster, as it will on a modern supercomputer like Titan. Any addition of accelerated code to the source must not interfere with the compilation process when using a standard F90 compiler or whenever building on architectures where accelerators are absent.
And, since my Ph.D. studies in Denmark cannot last for longer than three years, I am forced to care as much about chemistry, (so applying the program to molecular problems in organic chemistry or material sciences), as on the actual implementation of a given theoretical method. For this reason, it has been imperative that the accelerated code should be relatively easy to write (from scratch), port, extend (possibly by others), and maintain (most likely by others).
B: What steps did you take to familiarize yourself with OpenACC?
J: During a visit to OLCF in late 2012, we were advised to use the accelerator directives of OpenACC as an alternative to CUDA C. Upon returning home to Denmark, I then consulted the many tutorials and presentations that were available online already at that time. After attending an OpenACC school in Stuttgart later that year, the first parts of an accelerated code quickly came together.
B: How long did it take to get satisfactory performance?
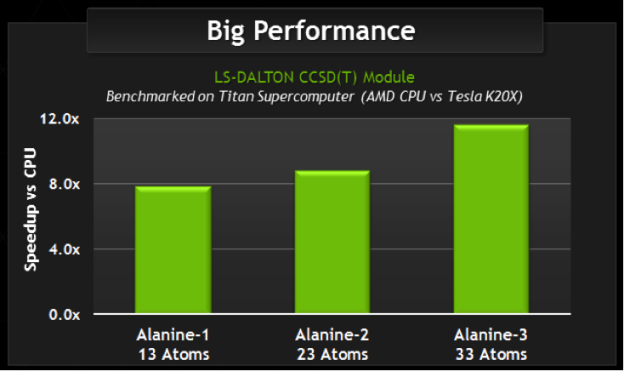
J: Despite spending next to no time writing an initial, fully synchronous OpenACC implementation of the method(s) in question, our developments were initially hampered by the fact that the implementations of OpenACC in the Cray and PGI compilers were still rather immature at that point (late 2012/early 2013). The compilers are now production ready and the code has since then developed to a more advanced state such that we are now extensively making use of newer features of the OpenACC 2.0 standard (nested parallelism, unstructured data regions, asynchronous wait operations, etc.) and dynamically choosing between different paths through the code at runtime, depending on what type of device we are executing the accelerated code on and how much device memory we have available on a given target device.
My ongoing goal is to extend the asynchronicity of the accelerated code to be able to run on multiple devices per compute node and document the implementation in full (alongside a lot more results).
B: What are the advantages of using OpenACC for domain scientists?
J: To me, the main advantage of OpenACC is the fact that any accelerated code making use of the standard will be based on original source code — regardless of whether this is written in FORTRAN, C, or C++ – making the implementation intuitively transparent and thus easier to maintain and extend. Furthermore, because the OpenACC programmer leaves most of the hard work to the compilers, and thus to the current and future developers of these, you are guaranteed that your code will execute on future architectures as well. Also, we have chosen to live by Amdahl’s law in our group, so while we began by porting the most compute intensive/expensive parts of our code, we are as such not bothered about a potential loss in performance from using OpenACC instead of CUDA C, as other parts of the code have already become new overall bottlenecks.
Finally, the fact that the same compiler may compile the same code (on the same architecture) with/without porting the code to the attached accelerators, by e.g. suppressing the accelerator directives for the non-accelerated build, we may directly evaluate the performance of our OpenACC implementation. Also, the (many) accelerator directives in our code are treated as mere comments to non-accelerating compilers, which make the efforts that we have to invest in making our program portable between different architectures negligible.
B: How has OpenACC impacted your research?
J: To me, as a PhD student, I’d say that OpenACC has impacted my research in two positive ways. First, being able to accelerate my code with minimum effort, saving time I would otherwise have spent on mastering a lower-level approach, has been not just a scientific accomplishment, but also a great personal satisfaction.
Furthermore, for me personally, adapting a new skill has been a lot of fun, and I’m convinced that the lessons learned (and the contacts made) will continue to prove beneficial to me in the future. I’m the first to admit that I don’t possess the same kind of low-level knowledge of GPUs or accelerators in general that a specialist has, but I’m confident that I can now (i) distinguish good accelerated code from bad and (ii) fairly easily make at least an initial attempt at accelerating a compute intensive code based on my knowledge of OpenACC alone.