Increasingly, computational chemistry researchers use GPUs to push the boundaries of discovery. This motivated Christopher Cooper, an Instructor at Universidad Técnica Federico Santa María in Chile, to move to a Python-based software stack.
Increasingly, computational chemistry researchers use GPUs to push the boundaries of discovery. This motivated Christopher Cooper, an Instructor at Universidad Técnica Federico Santa María in Chile, to move to a Python-based software stack.
Cooper’s recent paper, “Probing protein orientation near charged nanosurfaces for simulation-assisted biosensor design,” was recently accepted in J. Chemical Physics.
Brad: Can you talk a bit about your current research?
Christopher: I am interested in developing fast and accurate algorithms to study the effect of electrostatics in protein systems. We use continuum models to represent the solvent around the protein (water with salt) via the Poisson-Boltzmann equation, and solve it with an accelerated boundary element method. We call the resulting code PyGBe, which is open-source software with an MIT license, and is available to download via the Github account of the research group where I did my Ph.D. at Boston University.
An important part of this effort has been related to open science and reproducibility. Not only is PyGBe open source, but the figures in our papers are available through Figshare under a Creative Commons CC-BY license. In the Figshare repository, we also have scripts that call PyGBe with the correct input parameters to reproduce the results of our paper with a single command. In fact, Professor Barba’s research group has made a consistent effort for reproducibility and open science for a few years now.
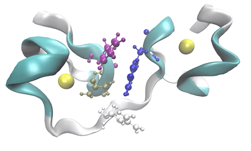
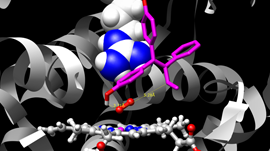
An interesting application we’ve been working on lately is label-free nanoscale biosensors. These are devices that detect antigens with very high sensitivity and selectivity. The antigen is sensed when it binds to a ligand that is adsorbed on the sensor, which needs to be properly oriented. We are using our model to find the best conditions to obtain favorable ligand orientations, and that way help the development of better sensors. This work has led to very interesting conversations with people outside our field, like experimentalists that fabricate these devices, which have been fun and challenging.
B: What tools do you use to develop Python code for GPUs?
We develop Python applications for GPUs using PyCUDA. This is a very nice combination because Python makes it very easy to move data around and sort it such that we get the most out of the GPU in the computationally intensive parts.

B: Why did you start looking at using NVIDIA GPUs?
C: I started working with NVIDIA GPUs over five years ago while a graduate student in Professor Lorena Barba’s group. We wanted to develop an efficient Poisson-Boltzmann solver that would be able to run on a local workstation using a boundary element method. It turns out that the most time consuming part of the algorithm can be formulated as an N-body type problem, which matches very well to the GPU architecture. Moreover, it was important for us to have a good double-precision performance which made NVIDIA GPUs the natural choice.
B: What is your GPU Computing experience and how have GPUs impacted your research?
C: I have been involved with CUDA in several ways. In 2011, I taught a month-long workshop on GPU computing with CUDA at Universidad Técnica Federico Santa María in Valparaíso, Chile (which was recently named a GPU Education Center). I also presented the early stage of my research in NVIDIA’s booth at the SC11 conference in Seattle, Washington.
GPUs and CUDA have had a huge impact on my research. For example, in the work on ligand orientation we sampled the free energy of antibodies near charged surfaces for nearly 1,000 different configurations – this would have taken over a week with the CPU version of the code, but we had the results in under a day!
B: Can you share some performance results?
C: The results in Figure 3 compare the time taken to calculate the solvation energy of a Lysozyme for a varying number of boundary elements, using one NVIDIA Tesla C2075 GPU and one Intel Xeon X5650 CPU.
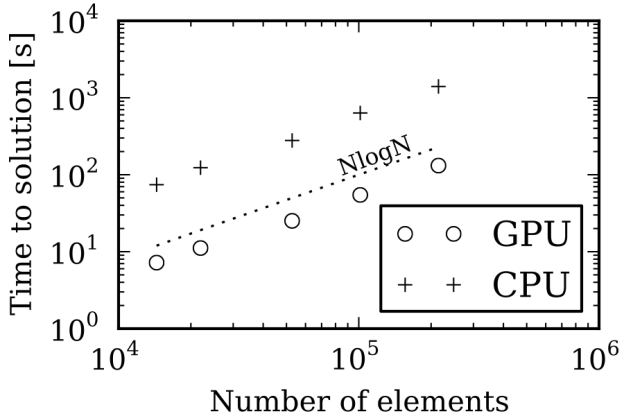
The NlogN scaling is expected because of the treecode algorithm, and you can see a 10x speedup between GPU and CPU.
B: How are you using GPUs?
C: We boil down a matrix-vector multiplication into an N-body problem which we accelerate with a treecode algorithm. The treecode performs a direct N-body calculation for near-by interactions, and approximates the far-field. The way that each evaluation point interacts with the near and far field bodies is independent, and we do them in parallel on the GPU.
B: What NVIDIA technologies are you using?
C: Besides GPUs, I take advantage of CUDA. One of the goals of the project was to develop a desktop-level, straightforward tool that would be easy to use for someone without a computer science background. This is why we did most of the development in Python, and interface with CUDA using PyCUDA.
B: What types of challenges have you encountered in your research?
C: The software development was a big challenge. Even though the treecode algorithm maps very well to the GPU architecture, the implementation was not straightforward and we had to be very careful in order to get the most from the GPU.

In our case, the key was to arrange the data in such a way that points of our N-body problem that were near in space, were also close by in memory. That way, we take advantage of the bandwidth, and make efficient use of the shared memory.
Arranging data gave us important speedups, but it was not the end of the story. We checked the CUDA code line by line to recognize bottlenecks and find better ways to perform each operation. The CUDA fast math instructions were very useful at this point! It was an exhausting process, but it definitely paid off.
Another challenge was linking the computational work that we were doing to a real application where it could have an impact. Once we saw an opportunity to study biosensors with PyGBe, we had to quickly educate ourselves in the topic, to understand specifically where our code would have the largest impact. In the end, some of the most interesting conversations I’ve had on this were with experimentalists that are not part of the computational world.
B: What’s next?
C: There are many interesting lines of work that come out from here. On the biosensor application, we looked at the orientation of antibodies near the sensor surface. Even though antibodies are widely used as ligands, there is active research engineering new ligands to enhance the sensitivity even more.
We plan to study the orientation of those new ligands adsorbed on the sensor surface using PyGBe, which can be very useful to help find better ligands.
I think the future of numerical modeling in biophysics will be dictated by how realistic and fast our simulations can get. There are quite a few number of ways to do biomolecular modeling (Molecular Dynamics, Generalized Born, Poisson-Boltzmann, etc.), with various levels of detail. The questions are: how realistic do we need to be? and, how long are we willing to wait for an answer?
The model in PyGBe has limitations, and we are interested in developing our code further towards getting more realistic simulations. In that context, memory constraints allow us to model only one or two biomolecules at the same time (depending on the size); however, in real life, biomolecules are interacting with many other molecules that are immersed in the same solvent. How do they interact? What is the effect of this crowding? To answer these questions, we need to develop a large-scale distributed memory version of PyGBe. Perhaps it won’t be easy to use like the original version, but it will allow us to add realism to our simulations.
B: What impact do you think your research will have on this scientific domain?
C: The biosensor application that we have been working on is a nice example of this. Most developments in this domain are guided by experiments and rely on trial-and-error. These biosensors are nanoscale devices, which are difficult and expensive to manipulate. An analytical tool like PyGBe can support those efforts and perhaps limit the number of trials to obtain a satisfying result.
B: Where do you obtain technical information related to NVIDIA GPUs and CUDA?
C: NVIDIA GPUs and CUDA have been around for a while now so information is readily available online. A good source to keep up with the latest is CUDA Zone in the NVIDIA Developer Zone. Also, when I started coding in CUDA I found the book “Programming Massively Parallel Processors,” by David Kirk and Wen-mei Hwu very useful.